Although RAS mutations constitute some of the most common genetic alterations in patients with adenocarcinomas, efforts to target the RAS oncoprotein have not proven fruitful, not only in NSCLC but across all cancers. In fact, researchers have been searching for an effective RAS inhibitor for more than 3 decades, leading many to believe that RAS is “undruggable.”
“Although the RAS gene is the oldest driver oncogene in lung cancer, RAS-targeted therapy has been challenging,” acknowledged IASLC President Tetsuya Mitsudomi, MD, of the Kindai University Faculty of Medicine in Japan.
Now, thanks to a better understanding of RAS disease biology and the emergence of novel technologies, advances are finally being made in this space, as detailed during the Education Session (ES28) focused on “Targeting KRAS.” Dr. Mitsudomi began the session by reviewing the biology of KRAS and various strategies for inhibiting its oncogenic activity when mutated.
The Basics of RAS Activation
The RAS gene encodes a protein that cycles between an inactive conformation bound by guanosine diphosphate (GDP) and an active conformation bound by guanosine triphosphate (GTP). When activated, RAS interacts with at least 20 different effector proteins, prompting them to elicit downstream signaling cascades.1 These downstream effectors include RAF, which activates the MAPK/ERK pathway to result in cell proliferation; RalGDS, which activates GTPases that mediate cell transformation and cytoskeletal reorganization; and PI3K, which activates the Akt pathway that promotes cell survival, growth, and migration.
The RAS gene family consists of three variants: HRAS, KRAS, and NRAS. Activating point mutations within these genes typically occur within codons 12, 13, or 61; however, activating mutations can also sometimes arise in codons 59, 117, or 146 within KRAS and NRAS.2 In general, all of these mutations disrupt RAS GTP hydrolysis, ultimately leaving RAS in a state of constitutive activation, according to Dr. Mitsudomi.
Different RAS gene variants predominate in different cancer types.3 KRAS is the predominant variant mutated in lung adenocarcinoma (found in 32% of cases), colorectal adenocarcinoma (found in 41% of cases), and pancreatic ductal adenocarcinoma (found in 86% of cases), whereas NRAS predominates in melanoma (found in 29% of cases) and HRAS in head and neck squamous cell carcinoma (found in 5% of cases) and urothelial carcinoma (found in 4% of cases).
There are also differences in amino acid substitutions according to tumor type.3 Most KRAS mutations in lung cancer occur in codon 12 and typically feature the glycine to cysteine (G12C) substitution, which arises from a G-to-T transversion driven by aromatic hydrocarbons contained in tobacco smoke. In contrast, the KRAS G12D substitution, arising from a G-to-A transition predominates in colorectal cancer and pancreatic ductal adenocarcinoma.
In in vitro studies, the G12C mutation causes impaired GAP-mediated hydrolysis of GTP, although it leaves the intrinsic GTPase activity of RAS nearly intact unlike other RAS mutations.4
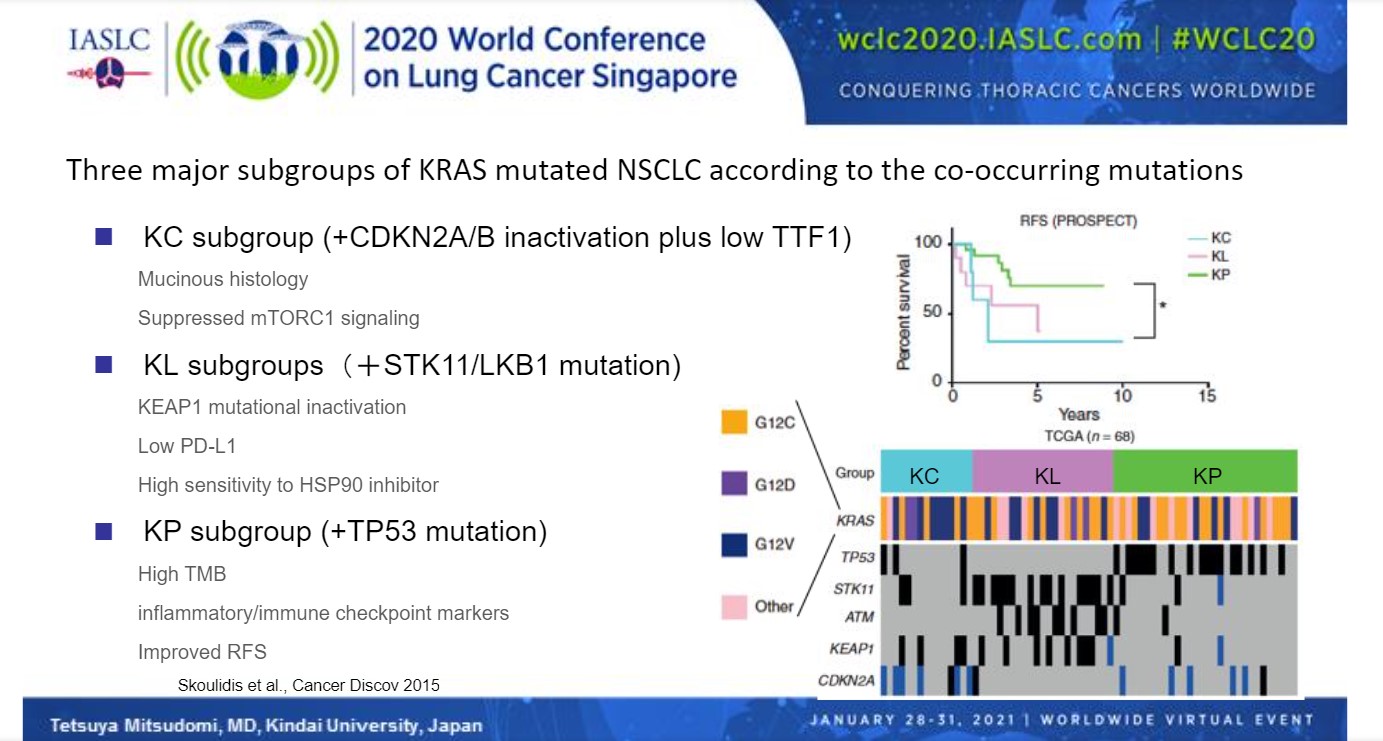
Clinically, there appears to be no major difference in prognosis based on different KRAS mutations.5 However, the presence of other concomitant mutations in other genes can alter the clinical picture. In fact, subgroups of KRAS-mutated NSCLC based on co-occurring mutations have been proposed.6 “These subtypes may reflect different biology and thus different therapeutic vulnerabilities,” said Dr. Mitsudomi.
The KC subgroup features KRAS mutations plus CDKN2A/B inactivation and low TTF1 expression, and is characterized by invasive mucinous histology, suppressed mTORC1 signaling, and a high likelihood of disease recurrence.6 The KL subgroup features KRAS mutations plus STK1/LKB1 mutations, and is characterized by KEAP1 mutational inactivation, low PD-L1 expression, high sensitivity to HSP90 inhibitors, and an intermediate risk of disease recurrence. The KP subgroup features TP53 mutations in addition to KRAS mutations, and is characterized by high tumor mutation burden, high levels of inflammatory or immune checkpoint markers, and a low likelihood of disease recurrence (Fig 1).
RAS-Targeted Drug Development
“One of the reasons for difficulty in targeting KRAS is the fact that not all KRAS-mutated tumors are dependent on the expression of mutant RAS for their survival,” Dr. Mitsudomi stated.
Tumor cells that proliferate independent of mutated KRAS are characterized by a mesenchymal phenotype.7 “This means that even if we have very nice KRAS inhibitors, we cannot cure the cancers with a mesenchymal phenotype,” said Dr. Mitsudomi.
Several strategies have been pursued to inhibit KRAS mutated lung cancer, including blocking KRAS association with the membrane, inhibition of downstream effectors, inhibition of selected proteins to result in synthetic lethality, and direct KRAS blockade.3 Unfortunately, most of these efforts have failed.
For example, early research focused on inhibiting RAS binding to the inner plasma membrane, which is essential for activating downstream signaling. Efforts sought to disrupt some step of the prenylation process required for membrane localization using a variety of compounds (eg, lonafarnib, tipifarnib, salirasib), but none has yet proved successful.8
Inhibition of downstream RAS effectors including RAF or MEK with agents such as sorafenib, selumetinib, and trametinib has also largely failed, primarily due to the many feedback mechanisms modulating this pathway.9 “If one of these molecules is inhibited, it will also inhibit negative feedbacks to ERK, RAF, or RAS itself, resulting in reactivation of the pathway,” Dr. Mitsudomi explained.

A number of genes that, when silenced, cause synthetic lethality in combination with KRAS mutations have been identified through shRNA screening, chief among them CDK4 (Fig 2). Abemaciclib, a CDK4/6 inhibitor, was recently compared with erlotinib in the phase III JUNIPER trial in patients with a detectable KRAS mutation who previously failed platinum-based therapy.10 Although abemaciclib significantly improved median progression-free survival compared with erlotinib (3.62 vs 19.1 months; HR 0.583, 95% CI [0.470, 0.723]; P < 0.0001), use of the CDK4 inhibitor showed no improvement in median overall survival, the primary endpoint (7.43 vs. 7.82 months; HR 0.968, 95% CI [0.768, 1.219]; P = 0.7714).
In other work, “an attempt to directly compete with GTP, as in the case of receptor tyrosine kinase inhibition, is difficult because of the very high affinity between RAS and GTP,” Dr. Mitsudomi explained.
To circumvent these many challenges, other novel strategies are being investigated, including disruption of SOS1-RAS interactions; inhibition of SHP2, an enzyme that modifies RAS to activate the MAPK pathway; blockade of guanine nucleotide-exchange factors (GEFs) that mediate upstream RAS signaling; and use of immunotherapies to target mutated KRAS proteins. However, one of the most promising new strategies being explored is KRAS G12C inhibition. KRAS G12C inhibitors irreversibly bind to the mutated cysteine residue within the protein and occupy the pocket near the switch II region of GDP-bound KRAS, essentially locking the mutated protein in an inactive state.
Subsequent talks during the “Targeting KRAS” Education Session focused on the development of KRAS G12C inhibitors, as well as some of the other novel therapies. These talks can be accessed through the On-Demand recordings, available through the virtual platform. Registration is ongoing for the next 60 days at wclc2020.iaslc.org.
References
- Erijman A, Shifman JM. RAS/effector interactions from structural and biophysical perspective. Mini Rev Med Chem. 2016;16(5):370-375.
- Smith G, Bounds R, Wolf H, Steele RJ, Carey FA, Wolf CR. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours – implications for personalised cancer medicine. Br J Cancer. 2010;102(4):693-703.
- Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533-552.
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res. 2015;13(9):1325-1335.
- Mascaux C, Iannino N, Martin B, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92(1):131-139.
- Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5(8):860-877.
- Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489-500.
- Vasan N, Boyer JL, Herbst RS. A RAS renaissance: emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin Cancer Res. 2014;20(15):3921-3930.
- Kitai H, Ebi H. Key roles of EMT for adaptive resistance to MEK inhibitor in KRAS mutant lung cancer. Small GTPases. 2017;8(3):172-176.
- Goldman JW, Mazieres J, Barlesi F, et al. A randomized phase III study of abemaciclib versus erlotinib in patients with stage IV non-small cell lung cancer with a detectable KRAS mutation who failed prior platinum-based therapy: JUNIPER. Front Oncol. 2020;10:578756.