






New insights into the types of EGFR mutations that can arise in NSCLC may help guide decisions about the most appropriate targeted drugs for individual patients.
A study by Jacqulyne Robichaux, PhD, of MD Anderson Cancer Center and colleagues (MA13.07) that used patient data, cell lines, and computerized models to analyze EGFR mutations found that the mutations can be divided into four functional subgroups, each resistant or sensitive to specific approved or experimental drugs. The results highlight not only the importance of genetic sequencing of tumors in patients with NSCLC both at diagnosis and after drug resistance has developed, but also the need for oncologists to understand the treatment sensitivities associated with each type of mutation.
“These findings show by classifying EGFR mutations based on their structure and function, rather than simply based on their exon, we are better able to identify the most appropriate EGFR inhibitors for patients with atypical EGFR mutations,” said the study’s lead author, Dr. Robichaux.
Dr. Robichaux and her colleagues embarked on the study because, despite advances such as the novel tyrosine kinase inhibitor (TKI) osimertinib—which has greatly improved outcomes for those patients with metastatic NSCLC harboring classical and T790M EGFR mutations—patients with atypical EGFR mutations do not uniformly experience good responses to EGFR inhibitors, indicating the need for more investigation.
Study Design and Findings
The researchers conducted a retrospective analysis of data from MD Anderson’s GEMINI database and Moffitt Cancer Center, evaluating the clinical outcomes of patients with atypical EGFR mutations who had been treated with different EGFR TKIs. The team found that 246 patients with classical EGFR mutations had a median progression-free survival (PFS) of 15 months, compared with 8 months for 113 patients who present atypical EGFR mutations (HR 2.0, p ˂ 0.0001).
To identify subgroups, the researchers used information from numerous databases to study 11,619 patients with NSCLC who had EGFR mutations. By using in silico mutational mapping, they determined the location of atypical mutations within the protein and predicted their impact on the structure of the drug-binding pocket. In addition, the team generated a panel of >70 Ba/F3 cell lines that expressed atypical mutations which occurred in more than 1% of patients and screened them against a panel of EGFR TKIs.
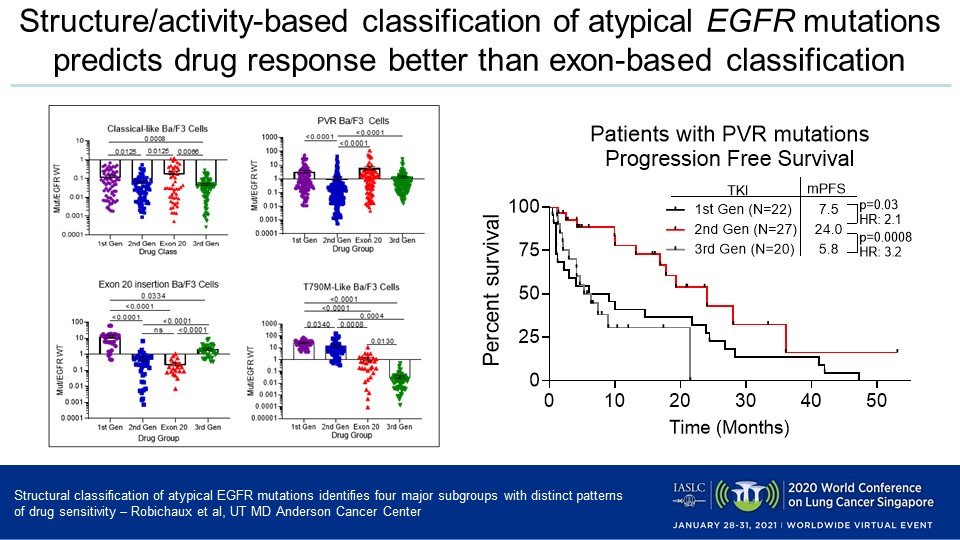
Of the patients analyzed, 67% had classical mutations, 31% atypical aberrations, and 2% a combination of mutations, Dr. Robichaux reported. Her team determined that atypical EGFR mutations are more diverse than classical ones and primarily occur in exons 18 and 20, and that EGFR mutations can be categorized into four distinct groups which respond differently to the distinct generations of EGFR inhibitors.
The four groups are:
- Classical-like: The EGFR mutations in this most common subset are distant from the ATP drug-binding pocket and sensitive to first-, second-, and third-generation inhibitors.
- T790M-like: These mutations in the hydrophobic core are sensitive to third-generation EGFR inhibitors but resistant to first- and second-generation inhibitors, irrespective of co-occurring mutations. A subset of this group included tertiary mutations that are resistant to EGFR inhibitors but sensitive to PKC and ALK inhibitors.
- Exon 20 insertions: These mutations, which occur on the back side of the alpha C helix and cause significant reduction in drug-binding pocket volume, are highly resistant to most classical EGFR inhibitors but sensitive to novel exon 20-specific inhibitors.
- ATP binding-pocket volume-reducing (PVR) mutations: These alterations appear on the interior of the ATP drug-binding pocket or C-terminal end of the alpha C helix and reduce the overall pocket volume. They are resistant to third-generation inhibitors, but sensitive to quinazoline-based, second-generation inhibitors such as afatinib and poziotinib.
The team demonstrated the clinical implications of these subgroups and according to the retrospective analysis, patients with PVR mutations had a significantly longer median PFS when treated with a second-generation inhibitor (24 months) versus a first-generation inhibitor (9.1 months, HR = 2.12, p = 0.016) or a third-generation member of the class (6.0 months, HR = 3.50, p < 0.0001). Among those patients were three who acquired PVR mutations after receiving first-line treatment with osimertinib, all of whom experienced clinical benefit after taking second-generation EGFR inhibitors.
“Together, these subgroupings may better guide future treatment approaches and clinical trial designs for patients with EGFR mutant NSCLC,” Robichaux said. “Further, these findings suggest that a structure-based approach may be used to predict targeted therapy sensitivity in oncogenes with diverse driver mutations.”
Discussant Jin-Yuan Shih, MD, of National Taiwan University Hospital, called the classification system “a novel approach.” He noted that there are “wild variations of drug sensitivity among each subgroup” and that it is not yet known how to specifically match drugs to atypical EGFR mutations. Dr. Shih suggested that more data be collected about clinical treatment outcomes and acquired resistance in these patients, and that a global database of atypical EGFR mutations be created.